ООО "ПРОПИОНИКС"
| пн-пт с 09:00 до 18:00 | |
Метаболически ассоциированная жировая болезнь печени, триптофан и микробиом
Неалкогольная жировая болезнь печени, триптофан, микробиом и иммунитет


Взаимодействия между метаболизмом триптофана, кишечным микробиомом и иммунной системой как потенциальные факторы неалкогольной болезни печени (НАЖБП) и метаболических заболеваний
Резюме
Распространенность неалкогольной жировой болезни печени (НАЖБП) растет и, следовательно, является ее бременем, поскольку НАЖБП является фактором риска цирроза и ассоциируется с другими метаболическими заболеваниями, такими как диабет II типа, ожирение, дислипидемия и атеросклероз. Связь между этими кардиометаболическими заболеваниями заключается в состоянии воспаления низкой степени тяжести с более высокими уровнями цитокинов и С-реактивного белка, обнаруживаемыми у лиц с НАЖБП, ожирением и диабетом II типа. Возможной терапевтической мишенью для уменьшения этого состояния слабого воспаления является метаболизм незаменимой аминокислоты триптофана. Его три основных метаболических пути (кинурениновый путь, индольный путь и путь серотонина/мелатонина) приводят к образованию таких метаболитов, как кинуреновая кислота, ксантуреновая кислота, индол-3-пропионовая кислота (IPA) и серотонин/мелатонин. Кинурениновый путь регулируется индоламин 2,3-диоксигеназой (IDO), ферментом, который активируется провоспалительными молекулами, такими как INF, IL-6 и ЛПС. Более высокая активность IDO связана с усилением воспаления и фиброза при НАЖБП, а также с повышением уровня глюкозы, ожирением и атеросклерозом. С другой стороны, повышенные концентрации метаболитов индольного пути, регулируемые кишечным микробиомом, по-видимому, приводят к более благоприятным исходам. В этом описательном обзоре обобщены взаимодействия между метаболизмом триптофана, микробиомом кишечника и иммунной системой как потенциальными факторами кардиометаболических заболеваний при НАЖБП.
1. Введение
Неалкогольная жировая болезнь печени (НАЖБП) поражает >25% населения во всем мире [1]. Как показывают несколько моделей прогнозирования, заболеваемость НАЖБП и, следовательно, бремя этого заболевания в ближайшие годы будут еще больше увеличиваться, и это часто связывают с пандемией ожирения [2,3]. НАЖБП определяется как стеатоз печени, подтвержденный визуализацией или гистологией, и без признаков вторичной причины (например, вирусный или аутоиммунный гепатит, алкоголь или лекарства). Гистологически НАЖБП можно разделить на неалкогольную жировую болезнь печени (НАЖБП) и неалкогольный стеатогепатит (НАСГ), причем последний увеличивает риск прогрессирования до цирроза [4] и гепатоцеллюлярной карциномы [5]. Кроме того, НАЖБП и НАСГ связаны с целым рядом кардиометаболических состояний, таких как сахарный диабет 2 типа (СД2) и сердечно-сосудистые заболевания. Учитывая растущую распространенность НАЖБП и ее связь с широким спектром хронических заболеваний, срочно необходимо лучшее понимание патогенеза НАЖБП и ее потенциального прогрессирования до НАСГ и цирроза печени.
Хотя ожирение, по-видимому, является основной причиной НАЖБП [6], патогенез НАЖБП и особенно его прогрессирование в сторону НАСГ предлагается в качестве "модели множественного поражения", в которой взаимодействие между несколькими путями приводит к повреждению печени, воспалению и фиброзу [7,8]. В настоящее время основными установленными патофизиологическими путями при НАЖБП считаются липотоксичность, приводящая к апоптозу и, следовательно, провоцирующая воспалительную реакцию. Со временем непрерывная нагрузка липидами приводит к окислительному стрессу с конечным результатом - фиброгенезом [7]. Корреляция между НАЖБП и метаболическими заболеваниями, такими как СД2, ожирение и сердечно-сосудистые заболевания (например, гипертония, гипертриглицеридемия и атеросклероз), хорошо установлена [6,9,10]. В популяции лиц с положительным ультразвуковым исследованием на наличие стеатоза 67,5% страдали ожирением, у 68,2% была гипертония, а у 26,3% был диагностирован диабет [11]. И наоборот, у пациентов с сахарным диабетом 56% соответствуют критериям НАЖБП [12].
Кроме того, хорошо изучены новые факторы риска, такие как ось кишечник–печень, и это один из путей, связанных с воспалением печени при НАЖБП [13]. Ось кишечник–печень в настоящее время понимается как нарушение микробного состава кишечника и нарушение кишечного барьера, повышающее проницаемость кишечного барьера. Следовательно, это приводит к транслокации микробов и микробных продуктов, таких как липополисахарид (ЛПС) и другие метаболиты, в портальный кровоток, непосредственно воздействуя на печень [14,15,16]. ЛПС связывается с toll-подобным рецептором 4 (TLR4), активируя врожденную иммунную систему в печени, особенно клетки Купфера и инфильтрирующие моноциты/макрофаги [16,17]. В сочетании с активацией каскада MyD88/NF-κB (миелоидный фактор дифференцировки 88 (MyD88), который является важной молекулой-адаптером для большинства TLR, опосредует индукцию провоспалительных цитокинов через ядерный фактор kB (NF-kB) - ред.) связывание ЛПС с TLR4 индуцирует высвобождение провоспалительных цитокинов, таких как интерлейкин 6 (IL-6), IL-17, фактор некроза опухоли α (TNF-α) и интерферон-гамма (INF-γ), что в конечном итоге приводит к высвобождение фиброгенных факторов [8,18,19].
Микробиом кишечника, в дополнение к его симбиотическим функциям, регулирующим воспалительный и иммунологический тонус хозяина [20] и предотвращающим чрезмерный рост патогенных вредных микробов, таких как Clostridium difficile, все чаще сравнивается с эндокринным органом [21]. Здесь кишечный микробиом вырабатывает целый ряд метаболитов, которые взаимодействуют со специфическими рецепторами, изменяя фенотип хозяина. Фактически, в связи с новыми представлениями о патофизиологии НАЖБП и ее растущей распространенности международная консенсусная группа недавно предложила изменить термин НАЖБП на метаболически ассоциированная жировая болезнь печени (МАЖБП или MAFLD) и скорректировать диагностические критерии с учетом признаков стеатоза печени в сочетании с ожирением или СД2 или признаков метаболической дисфункции [22]. Ключевым путем, представляющим интерес в этом контексте, является выработка метаболитов триптофана либо хозяином, либо кишечным микробиомом [23].
Более низкие уровни триптофана и повышенные ферменты, связанные с триптофаном (например, индоламин 2,3-диоксигеназа 1 и 2 (IDO1 и IDO2) и триптофан-2,3-диоксигеназа (TDO)) и нижестоящие метаболиты были связаны с усилением метаболического воспаления и фиброза [24,25]. При НАЖБП, в частности, метаболиты индольного пути, превращаемые кишечным микробиомом, как было показано, уменьшают воспаление через путь NF-κB, а некоторые другие метаболиты, как было показано, снижают выработку цитокинов, таких как IL-22, и модулируют врожденную иммунную систему [26]. Эти пути имеют потенциальное значение, поскольку нарушение метаболизма триптофана может быть относительно легкой терапевтической мишенью в контексте НАЖБП/НАСГ либо с помощью диетических стратегий, пробиотиков (содержащих микробиоту, которая метаболизирует триптофан), либо полученных из микробиоты метаболитов триптофана с благоприятными свойствами (концепция, называемая постбиотиками). В этом обзоре мы представляем обзор физиологии метаболизма триптофана, рассматривая как метаболизм хозяина, так и метаболизм микроорганизмов. Из-за внутренней связи метаболитов триптофана и связанных с ними ферментов с микробиомом кишечника и иммунной системой, мы предполагаем, что нарушения в путях триптофана способствуют развитию НАЖБП, а также кардиометаболическим состояниям, которые, как считается, провоцируют НАЖБП. Это представляет большой интерес, поскольку НАЖБП и кардиометаболические заболевания являются чрезвычайно распространенными состояниями. Поэтому лучшее понимание физиологии триптофана, нарушений метаболизма триптофана из-за метаболической дисрегуляции и его потенциального влияния на метаболические заболевания имеет жизненно важное значение.
2. Физиология метаболизма триптофана и метаболитов триптофана
2.1. Поступление, всасывание и выведение триптофана
Триптофан является одной из девяти незаменимых аминокислот и в относительном изобилии содержится в индейке, курице, молоке, тунце, орехах и бананах. Среднее потребление триптофана у взрослых составляет около 900–1000 мг/день, тогда как рекомендуемое суточное потребление составляет около 3,5–6,0 мг/кг [27]. Триптофан вместе с фенилаланином и тирозином называют ароматической аминокислотой из-за боковой цепи бензольного кольца, которая в триптофане специфически состоит из индола [28].
 Прим. ред.: индольное кольцо (рис. слева) состоит из бензольного кольца, сплавленного с пятичленным азотсодержащим пиррольным кольцом.
Прим. ред.: индольное кольцо (рис. слева) состоит из бензольного кольца, сплавленного с пятичленным азотсодержащим пиррольным кольцом.
Катаболизм триптофана включает три основных пути; кинурениновый путь, на который приходится около 90–95% метаболизма триптофана, серотонин/мелатониновый путь, на который приходится около 1–2% метаболизма триптофана, и 5% используются в индольном пути [27,29]. После приема внутрь большая часть белков и аминокислот всасывается в тонком кишечнике (см. рис. 1) [29]. Потребление аминокислот с пищей, по-видимому, не влияет на концентрацию аминокислот в цитозоле или крови, но потребление углеводов или белков изменяет доступность 5-гидроксииндолуксусной кислоты (5HIAA) в головном мозге, что свидетельствует о тканезависимом контроле концентрации триптофана [30].

Рисунок 1. Поглощение и метаболизм триптофана. Сокращения: 5-НТ, серотонин; АА0, нейтральная аминокислота; AhR, арильный углеводородный рецептор; GC, глюкокортикостероиды; IDO, идоламин-2,3-диоксигеназа; INF, интерферон; IL, интерлейкин; KYN, кинуренин; LPS, липополисахарид; MEL, мелатонин; PXR, прегнан X-рецептор; TDO, триптофан-2,3-диоксигеназа; TPH1, триптофангидроксилаза 1; Trp, триптофан.
Будучи нейтральной аминокислотой, триптофан поглощается ATB0+ (SLC6A14, симпортер, использующий 2Na+/1Cl-) и B0AT1 (SLC6A19, симпортер, использующий 1Na+) на апикальной мембране тонкой кишки и выводится в портальную циркуляцию через базолатеральную мембрану с помощью TAT1 (SLC16A10, унипортер) или LAT1-4F2hc (SLC7A5-SLC3A2, антипортер) и LAT2-4F2hc (SLC7A8-SLC3A2, антипортер) [31,32]. Эти транспортеры аминокислот также называются системой B+ (B0AT1/B0AT2), системой B0+ (ATB0+), системой L (LAT1-4F2hc/LAT2-4F2hc) и системой T (TAT1) [33]. Эти транспортеры часто должны связываться с другими молекулами, такими как ангиотензинпревращающий фермент 2 (ACE2), CD98/CD147 [34] или аминопептидаза N [35]. Считается, что ассоциация с другими белками необходима для встраивания транспортера в клеточную мембрану, для увеличения снабжения субстратом или для модуляции активности транспортера [34]. Факторы, влияющие на экспрессию этих рецепторов и белков, на сегодняшний день полностью не выяснены. После всасывания в кровоток триптофан становится единственной аминокислотой, которая связывается с альбумином (75–90%) [27,36]. Факторами, негативно влияющими на количество триптофана, связанного с альбумином, являются низкие концентрации альбумина, некоторые лекарства и неэтерифицированные жирные кислоты [37].
Те же переносчики аминокислот, что и в кишечнике, присутствуют и в почках. На люминальной щеточной границе проксимальных почечных канальцев наблюдается обильная экспрессия B0AT1, зависящая от коллекрина, в отличие от ACE2 в кишечнике. На базолатеральной мембране TAT1 часто располагается вместе с LAT2-4F2hc (SLC7A5-SLC3A2, антипортер) и выводит нейтральные аминокислоты во внеклеточное пространство [38,39]. Триптофан, который не всасывается в тонкой кишке, будет использоваться в качестве источника энергии для микробиоты в толстой кишке, индольного пути, синтеза серотонина и небольшой части внепеченочного пути кинуренина [29].
2.2. Кинурениновый путь
Кинурениновый (основной) путь – окисление и разрушение индольного кольца с образованием производных кинуреновой и антраниловой кислот.
Кинурениновый путь является основным катаболическим путем триптофана (см. Рисунок 2). После всасывания в кровоток около 90% триптофана метаболизируется с помощью IDO1, IDO2 и TDO в N-формилкинуренин [40]. Это первая и ограничивающая скорость стадия кинуренинового пути [30], приводящая к последующему образованию никотинамидадениндинуклеотида (NAD), кинуренина, кинуреновой кислоты (KA), ксантуреновой кислоты (XA), пиколиновой кислоты (PA) и антраниловой кислоты (AA).

Рисунок 2. Кинурениновый путь. Сокращения: IDO, индоламин-2,3-диоксигеназа; TDO, триптофан 2,3-диоксигеназа.
TDO в основном экспрессируется в печени, но также и в тканях головного мозга, и его активность усиливается кортикостероидами. Интересно, что некоторые виды бактерий, такие как Pseudomonas Aeruginosa, экспрессируют TDO [29]. По сравнению с IDO, TDO имеет более высокое сродство с триптофаном и преобразует исключительно триптофан [41]. Одно исследование показало, что некоторые метаболиты индольного пути являются ингибиторами TDO, но эти метаболиты не ингибируют IDO [42]. IDO имеет сродство с несколькими субстратами и экспрессируется по всему телу в эпителиальных и эндотелиальных клетках, а также в моноцитах, макрофагах и клетках гладкой мускулатуры сосудов [43]. Активность IDO1 сильно индуцируется INF-γ, хотя другие провоспалительные молекулы также связаны с более высокой активностью IDO, такие как INF-α, INF-β, IL-6 и ЛПС [43,44]. IDO-активность часто измеряют по соотношению кинуренин/триптофан. Тем не менее, это подлежит обсуждению, поскольку другие факторы, такие как активность TDO и количество триптофана, связанного с альбумином, не учитываются при применении этого соотношения [37]. IDO2 обладает низкой ферментативной активностью по сравнению с IDO1 и TDO. Регуляция и механизмы IDO2 полностью не выяснены, но, по-видимому, IDO2 обладает отличными от IDO1 свойствами [45,46].
Регуляция активности нижестоящих ферментов и эффекты нижестоящих метаболитов подробно рассмотрены в нескольких статьях [37,40,47,48,49]. Эти метаболиты связаны с широким спектром заболеваний, включая НАЖБП, диабет и сердечно-сосудистые заболевания, и будут обсуждаться далее в этом обзоре.
2.3. Индольный путь
Индольный путь – образование индольных производных, которые затем конъюгируются выводятся с мочой.
Неабсорбированный триптофан, полученный с пищей, транспортируется к толстой кишке, где самые высокие концентрации триптофана обнаруживаются в дистальном отделе толстой кишки. Здесь индольный путь регулируется кишечным микробиомом и приводит к различным индолам, таким как индол-3-ацетат, индол-3-пропионат и скатол (3-метилиндол) (см. Рисунок 3) [50]. Ферменты, участвующие в формировании этих метаболитов, вырабатываются различными кишечными бактериями, такими как Clostridium spp, Bacteroides spp и Peptostreptococcus spp. Полный обзор видов, связанных с разложением триптофана, был недавно сделан Roager et al. [51]. Однако превращение индола в индоксилсульфат происходит в печени и опосредуется цитохромом Р450 и сульфотрансферазой [52,53]. Измерение и концентрации этих метаболитов значительно различаются в зависимости от образца, в котором они измеряются (например, в ткани головного мозга, спинномозговой жидкости, слюне, плазме и фекалиях) [54]. Кинетика всасывания и распределения индола до конца не изучена.

Рисунок 3. Индольный путь. Сокращения: AraT, аминотрансфераза ароматических аминокислот; МАО, моноаминооксидаза.
Связующими факторами микробных метаболитов и воспаления являются несколько человеческих рецепторов, влияющих на иммунную систему, таких как арилуглеводородный рецептор (AhR) и прегнан Х-рецептор (PXR) [55]. Эти рецепторы связаны с воспалением и поэтому действительно могут опосредовать некоторые аспекты оси кишечник-печень.
Метаболиты, такие как триптамин (TRP), 3-метилиндол (3MI), индол-3-ацетальдегид, IA, индол-3-альдегид, индолакриловая кислота, кинуренин, KA, XA и 5-гидроксииндолуксусная кислота (5-HIAA) являются лигандами для AhR [56,57]. AhR экспрессируется по всему телу, но самые высокие уровни обнаруживаются в кишечнике, легких и коже, и было показано, что он регулирует иммунную систему с помощью нескольких механизмов [58], таких как регуляция дифференцировки клеток Th17 и Treg (см. 4) [59,60].

Рисунок 4. Взаимодействие метаболитов триптофана с рецепторами врожденного иммунитета. Сокращения: 5-HIAA, 5-гидроксииндолуксусная кислота; 3MI, 3-метилиндол; 2IA, индол-3-альдегид; 3IA, индол-3-ацетальдегид; AA, антраниловая кислота; IA, индол-3-ацетат, IAA, индолакриловая кислота; КА, кинуреновая кислота; KYN, кинуренин; TRP, триптамин; XA, ксантуреновая кислота.
Индол и индол-3-ацетамид являются антагонистами PXR [61], который наиболее обильно экспрессируется в печени. Этот внутриклеточный рецептор участвует в понижающей регуляции глюконеогенеза и метаболизма липидов, а также в иммунной регуляции путем уменьшения воспаления путем подавления пути NF-kB [62,63].
2.4. Путь серотонина и мелатонина
Третий путь разложения триптофана приводит к образованию серотонина и мелатонина, на долю которых приходится 1-2% от общего разложения. Большая часть серотонина (95%) вырабатывается в желудочно-кишечном тракте энтерохромаффинными клетками, где триптофан превращается в 5-гидрокситриптофан триптофангидроксилазой 1 (TPH1), стадией, ограничивающей скорость, и, следовательно, в серотонин декарбоксилазой ароматических аминокислот (см. рис. 5) [64]. Микробиом и ось кишечник–мозг, по-видимому, играют важную роль в регуляции синтеза серотонина. Известно, что некоторые виды кишечных бактерий продуцируют серотонин in vitro, но способствует ли это повышению уровня серотонина у млекопитающих, неизвестно [65]. У мышей без микробов наблюдается более низкая экспрессия TPH1 и, следовательно, более низкие уровни серотонина в толстой кишке, кале и сыворотке крови, что указывает на регулирующую роль микробиома кишечника в концентрации серотонина [66]. Однако лежащий в основе механизм регуляции влияния кишечного микробиома на серотонин остается неясным. Из желудочно-кишечного тракта серотонин транспортируется через тромбоциты к различным периферическим участкам, таким как печень и сердечно-сосудистая система [67]. Синтез серотонина также происходит в нескольких клетках, таких как иммунные клетки (например, Т-клетки, В-клетки, моноциты и макрофаги) и бета-клетки поджелудочной железы [61]. Действие периферического серотонина зависит от различных рецепторов, с которыми он способен связываться [68].
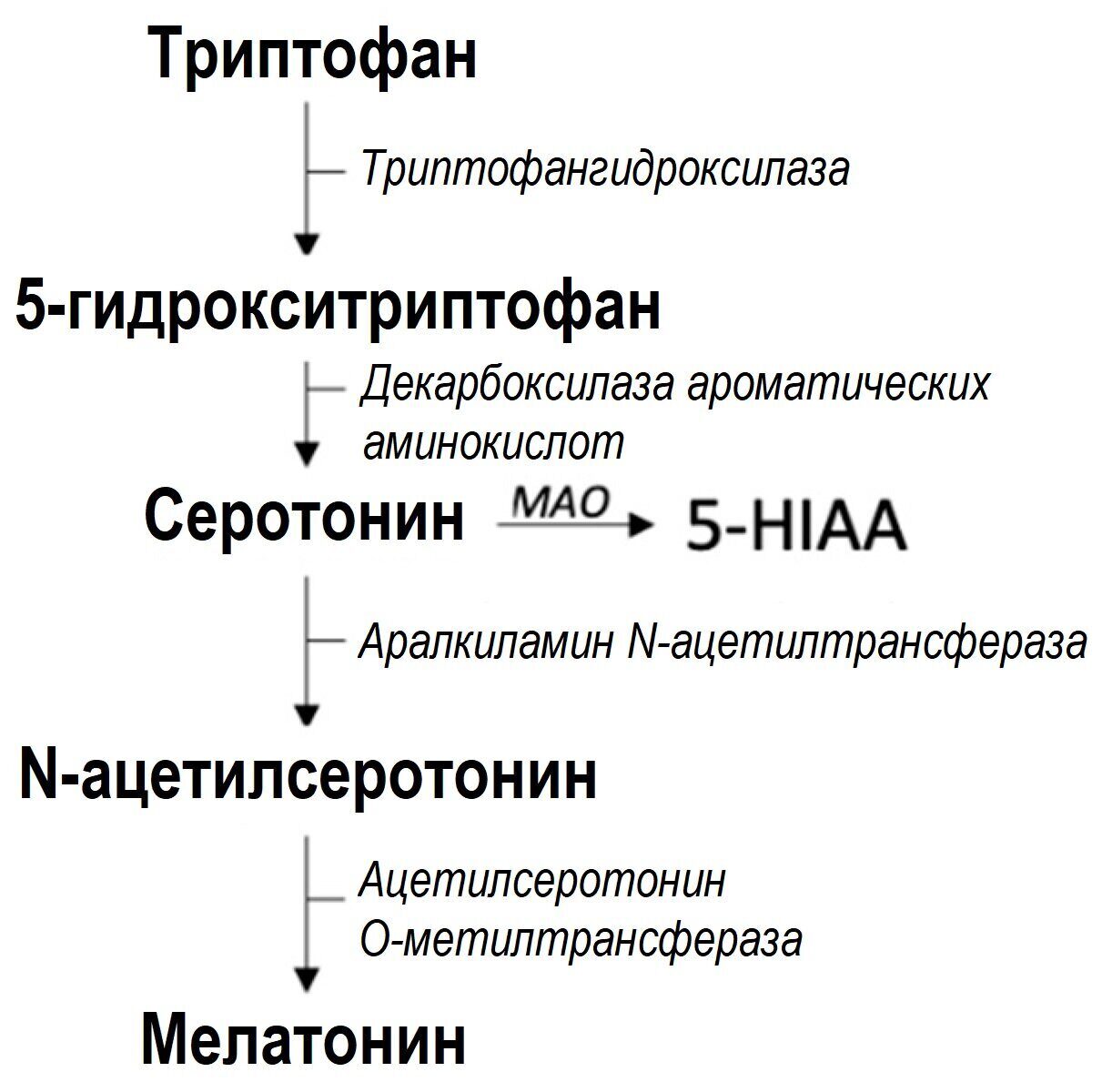
Рисунок 5. Серотониновый путь. Сокращения: 5-HIAA, 5-гидроксииндолуксусная кислота; МАО, моноаминооксидаза.
Остальная продукция серотонина происходит в ядрах шва, расположенных в стволе мозга. Поскольку триптофан не может преодолевать гематоэнцефалический барьер (ГЭБ), выработка серотонина в головном мозге зависит от поглощения триптофана через ГЭБ, что частично регулируется концентрацией других крупных нейтральных аминокислот (например, фенилаланина, валина) [69].
Последним этапом серотонинового пути является синтез мелатонина в шишковидной железе, а также в других местах по всему телу, таких как печень, желудочно-кишечный тракт и кожа [70]. Регуляция мелатонина сложна и выходит за рамки данного обзора.
3. Метаболизм триптофана при НАЖБП
3.1. Кинурениновый путь
Воспаление считается одним из центральных механизмов НАЖБП. Поскольку кинурениновый путь активируется воспалительными молекулами через IDO, он может играть важную роль в патогенезе НАЖБП. В образцах фекалий пациентов с НАЖБП уровни кинуренина действительно были повышены, а уровни триптофана снижены, что согласуется с более высокой активностью кинуренинового пути. Кроме того, микробиота была изменена по сравнению со здоровым контролем с обилием Collinsella, Acinetobacter и Actinomyces, что, в свою очередь, было связано с более высокими уровнями кинуренина [71].
Чтобы выяснить причинно-следственную связь этого наблюдения, современная литература в основном зависит от доклинических исследований. Мыши с нокаутом по IDO на диете с высоким содержанием жиров действительно демонстрировали более высокие уровни маркеров макрофагов и воспалительных молекул, таких как IFN-γ, IL-1β и IL-6, по сравнению с мышами с активностью IDO. Мыши с нокаутом IDO также имели повышенную экспрессию маркера фиброза TGF-β2, а гистология показала повышенный фиброз в образцах печени, предполагая, что активность IDO уменьшает воспаление и фиброз [72]. Напротив, у мышей на диете с дефицитом метионина и холина, которая вызывает стеатоз, при ингибировании IDO с помощью 1-метил-D-триптофана также снижалась экспрессия провоспалительных генов, кодирующих TNF-α и IL-1β, а также экспрессия TGF-β и альфа-гладкомышечного актина (α-SMA), что указывает на снижение фиброза. Таким образом, эти данные свидетельствуют о том, что снижение активности IDO может уменьшить воспаление и фиброз [73].
Когда стерильным (безмикробным) мышам трансплантировали образцы фекалий от пациентов с НАЖБП, наблюдалось повышение уровня кинуренина, а в образцах печени увеличивалось накопление внутрипеченочных липидов [71]. Эти результаты указывают на перекрестные взаимодействия между кишечным микробиомом и кинурениновым путем. Пероральное добавление кинуренина в сочетании с диетой с высоким содержанием жиров у мышей показало увеличение отложения жира в печени и усиление регуляции CYP1a1, CYP450 и Scd1, связанное с метаболизмом липидов в печени [74].
В заключение, повышенные концентрации кинуренина, по-видимому, стимулируют метаболизм липидов и внутрипеченочное отложение жира, тогда как роль IDO остается неясной.
3.2. Индольный путь
На несколько воспалительных механизмов при НАЖБП влияют микробные метаболиты индольного пути [75,76,77,78]. Потенциальным драйвером воспаления при НАЖБП является путь NF-kB, индуцируемый резидентными макрофагами печени (клетками Купфера) [18,19]. У грызунов пероральное введение индола после внутрибрюшинного введения ЛПС снижало уровни цитокинов IL-1β, IL-6 и IL-15, а также уровни NF-kB [79]. У мышей с ожирением и худых мышей пероральное употребление индола снижало экспрессию Cd68, что указывает на снижение накопления макрофагов [80], что согласуется с более ранними выводами о том, что индол снижает PFKFB3 в макрофагах, что подавляет их провоспалительное состояние [81].
Другое исследование показало, что IA (индол-3-уксусная кислота - IA вместо обозначения IAA, т.к. в статье путаница - IAA: индолакриловая кислота - ред.) уменьшала количество макрофагов в ткани печени и снижала уровни моноцитарного хемоаттрактантного белка 1 (MCP-1) и TNF-α у мышей с гепатостеатозом, вызванным диетой с высоким содержанием жиров [82]. У пациентов с ожирением более низкие уровни IA коррелировали с более высокими значениями КТ печени, указывающими на НАСГ. После рукавной гастростомы уровни IA в этой популяции увеличились, в то время как показатели КТ печени и уменьшение жира улучшились, что позволяет предположить, что IA обладает гепатопротекторными свойствами [83].
Наконец, было продемонстрировано, что IPA может ослаблять стеатоз печени за счет увеличения белков плотных контактов и, таким образом, укрепления желудочно-кишечного барьера, что приводит к снижению циркулирующих эндотоксинов и, следовательно, к снижению активации пути NF-kB посредством TLR4-активации [84]. Сегал и др. обнаружили, что более низкие уровни циркулирующей IPA у пациентов с ожирением без СД2 были обратно связаны с фиброзом в биоптатах печени. Кроме того, они показали, что обработка увековеченной линии звездчатых клеток печени человека (клетки LX-2) in vitro посредством TGF-β1 и IPA снижала активность звездчатых клеток, активируемых за счет снижения экспрессии мРНК COL1A2 и α-SMA, что связано с активацией звездчатых клеток печени [85].
Однако Лю и соавт. продемонстрировали, что у мышей, получавших хемокиновый лиганд 4 (CCL4), пероральное введение IPA приводило к увеличению экспрессии α-SMA и COL1A2, что фактически предполагает противоположный эффект in vivo. Интересно, что они также обнаружили, что CCL4-лечение уменьшало разнообразие микробиома кишечника, которое было обращено вспять после лечения IPA [86], что указывает на то, что IPA может модулировать изменения в микробиоме кишечника, вызванные воспалением.
Поскольку метаболиты индола продуцируются микробиотой кишечника, вышеупомянутые результаты подчеркивают потенциальную роль дисбиоза кишечника в этиологии НАЖБП, при этом несколько исследований указывают на изменения микробиома кишечника у животных и людей с НАЖБП [87].
3.3. Пути серотонина и мелатонина
У пациентов с НАЖБП повышенный уровень 5-HIAA, маркера серотонина, обычно используемого для скрининга нейроэндокринных опухолей, коррелировал с повышенным риском осложнений, связанных с печенью, таких как асцит и гепатоцеллюлярная карцинома [88].
In vitro антагонизм серотонина и последующее снижение уровня серотонина действительно приводили к снижению экспрессии мРНК фиброзных генов (α-SMA и COL1a1) и провоспалительных молекул (IL-1α и IL-8) [89]. В моделях мышей с НАЖБП блокирование серотониновых рецепторов трописетроном приводило к уменьшению стеатоза и фиброза в гистологических образцах печени [90]. Другое исследование показало увеличение рецептора серотонина 2A (HTR2A) в печени мышей с НАЖБП, в то время как у мышей с генетическим дефицитом рецептора HTR2A, а также при обработке антагонистом HTR2A, стеатоз печени снижался [83]. В связи с этим мелатонин изучался в нескольких контекстах, связанных с воспалением печени, например, путем индукции воспаления охратоксином [91]. У уток пероральный прием мелатонина приводил к снижению сывороточных уровней TNF-α, IL-1β, IL-6, а также к снижению экспрессии мРНК TLR4 [92]. Сходные результаты были получены у мышей [93], тогда как введение ЛПС крысам приводило к острой клеточной инфильтрации в биоптатах печени, которая купировалась пероральным введением мелатонина. Это также привело к снижению окисления липидов [94], что, в свою очередь, снизило прогрессирование НАЖБП [95]. У пациентов с НАЖБП, получавших 5 мг мелатонина два раза в день в течение 14 месяцев, наблюдалось значительное снижение уровня цитокинов IL-1, IL-6 и TNF-α по сравнению с группой плацебо, что еще раз указывает на способность мелатонина уменьшать воспаление [96].
В целом, серотонин, по-видимому, усугубляет фиброз и воспаление. Это также подтверждается повышением концентрации серотонина под действием селективных ингибиторов обратного захвата серотонина и последующим увеличением НАЖБП [97,98,99]. Несмотря на то, что мелатонин является нижестоящим метаболитом серотонина, его действие, по-видимому, противоположно действию серотонина, при этом мелатонин проявляет защитные свойства против фиброза и воспаления. Механизмы, которые могли бы определять сдвиг в сторону серотонина или мелатонина, на сегодняшний день неизвестны.
4. Триптофан и метаболические заболевания
НАЖБП тесно связана с метаболическими заболеваниями, такими как диабет, ожирение и атеросклероз. Связующим звеном этих заболеваний является гипервоспалительное состояние. Триптофан и его метаболиты по-разному ослабляют иммунную систему [100, 101, 102, 103, 104] и поэтому участвуют в нескольких патологических механизмах при метаболических заболеваниях.
4.1. Диабет
В большой финской когорте пациентов с СД2 более низкие уровни IPA и более высокие концентрации СРБ были обнаружены у пациентов с более низкой секрецией инсулина, что указывает на связь между вялотекущим воспалением и СД2, модулируемым IPA [105]. Другое исследование показало, что у пациентов с плохим гликемическим контролем концентрация триптофана была значительно ниже по сравнению с пациентами с умеренным диабетом, а также со здоровыми людьми. Они также обнаружили более высокое соотношение кинуренин/триптофан, оба результата указывают на более высокую активность IDO у пациентов с диабетом, который активируется воспалительными молекулами [106]. Другое исследование подтвердило результаты и даже обнаружило, что более высокое соотношение кинуренин/триптофан было связано с более высокой смертностью при СД2 [107].
На крысах было продемонстрировано, что многие ферменты, участвующие в кинурениновом пути, экспрессируются в β-клетках поджелудочной железы, но физиологическая роль не была обнаружена [108]. У мышей было обнаружено, что триптофан (и фенилаланин) избирательно связываются с GPR142, G-связанным рецептором с высокой экспрессией в клетках поджелудочной железы и кишечника, и поэтому могут модулировать секрецию инсулина и глюкагоноподобного пептида 1 (GLP-1) [109].
Хотя связь между воспалением, повышением активности IDO и СД2, по-видимому, установлена и находится в том же направлении, что и НАЖБП, лежащие в основе молекулярные пути, в которых различные метаболиты, такие как индолы, участвуют в контроле гликемии, и, следовательно, связь с микробиомом кишечника, также остаются неясными.
4.2. Ожирение
Подобная связь между вялотекущим воспалением и (избыточной) активностью кинуренинового пути, как и при НАЖБП, также обнаруживается у пациентов с ожирением. Как и у пациентов с СД2, концентрации триптофана оказались ниже у пациентов с ожирением по сравнению со здоровым контролем, а также у них было более высокое соотношение кинуренин/триптофан. С другой стороны, сывороточные концентрации IA*, IPA, ILA и индоксилсульфата были ниже у пациентов с ожирением, а повышенные концентрации СРБ и IL-6 были связаны с более низкими уровнями метаболитов индола [110]. Это еще раз указывает на то, что IDO-активность индуцируется провоспалительным состоянием, повышающим активность кинуренинового пути, тогда как образование метаболитов индола снижается.
Одно исследование показало, что блокирование AhR снижает ожирение у мышей на западной диете. Они предположили, что слабовыраженное воспаление увеличивает активность IDO, тем самым напрямую увеличивая концентрацию кинуренина. Поскольку кинуренин является агонистом AhR, чрезмерная стимуляция AhR более высокими концентрациями кинуренина может привести к ожирению [111]. Интересно, что у мышей с нокаутом IDO с ожирением наблюдается очевидный сдвиг в сторону индольного пути по сравнению с мышами дикого типа с повышенной активностью IDO в кишечнике и, что интересно, более высокими уровнями IA [112], что, возможно, указывает на зависимое от IDO образование индолов.
4.3. Атеросклероз
Роль кинуренинового пути при атеросклеротических заболеваниях была подробно рассмотрена [113, 114, 115]. Состояние вялотекущего воспаления при атеросклерозе характеризуется повышением уровня IL-6, индуктора IDO [116]. Еще раз, исследования показывают, что снижение триптофана и повышенное соотношение кинуренин/триптофан связано с тяжестью атеросклероза [113] и, таким образом, указывает на то, что повышенная активность IDO связана с этим состоянием вялотекущего воспаления.
Одно обсервационное исследование в когорте пациентов, перенесших каротидную эндартерэктомию, шунтирование конечности или ампутацию из-за ишемии, действительно показало, что соотношение кинуренин/триптофан в плазме было связано с прогрессирующим атеросклерозом, а также с более высоким риском послеоперационных сердечных осложнений. Однако они также продемонстрировали, что сывороточные концентрации IPA и индол-3-альдегида были ниже у пациентов с тяжелым атеросклерозом [117], что согласуется с исследованиями, предполагающими защитную роль индольного пути против воспаления.
В другом исследовании изучалось влияние антибиотиков на развитие атеросклероза у мышей. Введение антибиотиков привело к уменьшению количества видов Bacteroidetes и Clostridia, оба из которых связаны с метаболизмом триптофана в кишечнике, и они продемонстрировали, что сокращение этих видов было связано с уменьшением метаболитов триптофана, полученных из кишечника [51,118]. Более того, они продемонстрировали, что прием триптофана после антибиотиков может уменьшить размер поражения аорты у этих мышей, хотя связь с атеросклерозом не была значимой [118]. Это демонстрирует, что возмущения в микробиоме кишечника изменяют выработку метаболитов триптофана, и это оказывает измеримое влияние на сосудистые заболевания.
Важно отметить, что из-за широкого спектра метаболических заболеваний существует несколько других состояний, которые имеют общие этиологические факторы с НАЖБП и также связаны с метаболическими заболеваниями. Взаимодействие между метаболизмом триптофана и такими заболеваниями еще предстоит выяснить. Например, в случае обструктивного апноэ во сне (OSA) есть несколько интересных работ, касающихся взаимосвязи между метаболизмом триптофана и OSA [119,120]. Тем не менее, необходимы дальнейшие исследования, чтобы установить потенциальные корреляции и основные механизмы.
5. Выводы и перспективы на будущее
Метаболизм триптофана представляет собой сложную сеть микробных метаболитов человека и кишечника, взаимодействующих с несколькими физиологическими и патологическими процессами. Степень и последствия этих взаимодействий полностью не выяснены, но, как описано в этом обзоре, они представляются жизненно важными для иммунологического ответа, фиброза, гликемического контроля, метаболизма липидов и гормонального гомеостаза. В спектре кардиометаболических заболеваний, по-видимому, наблюдается дисбаланс в регуляции различных (кишечных и эндогенных) путей после деградации триптофана. Это может быть связано с повышенной активностью кинуренинового пути, на что указывает более высокая активность IDO и повышенные концентрации связанных нижестоящих метаболитов, что связано с усилением воспаления и фиброза и, следовательно, связано с метаболическими заболеваниями, такими как НАЖБП.
Кроме того, было продемонстрировано, что активность IDO является ключевым регулятором в этом процессе и что существует активный сдвиг, увеличивающий метаболиты кинуренина и уменьшающий метаболиты индола. Однако показано, что индольный путь обладает противовоспалительными свойствами, особенно при НАЖБП и атеросклерозе. Следовательно, повышение концентрации метаболитов по этому пути представляет терапевтический интерес.
Этот дисбаланс также заметен в пути серотонина, где серотонин, по-видимому, усугубляет НАЖБП. Добавление мелатонина уменьшает воспаление и фиброз.
Таким образом, мы предполагаем, что при НАЖБП баланс между путями IDO, индола и серотонина был смещен в сторону провоспалительных последующих действий метаболизма триптофана. Восстановление этого баланса (например, с помощью манипуляций с микробиотой) может стать важным шагом вперед для предотвращения прогрессирования НАЖБП в сторону НАСГ и ослабления связи между НАЖБП, СД2 и сердечно-сосудистыми заболеваниями при ожирении. С этой целью необходимы более фундаментальные исследования для разработки терапевтических стратегий, уменьшающих влияние триптофана на состояние с низкоуровневого воспаления при метаболических заболеваниях и тем самым, возможно, уменьшающих сердечно-сосудистые и метаболические осложнения.
- Микробиом кишечника человека и заболевания печени: от корреляции к причинно-следственной связи
- Связь микробиоты кишечника и заболеваний печени
- НАЖБП / НАСГ & микробиом кишечника
- Влияние микробиома на неалкогольную жировую болезнь печени (НАЖБП)
- Микробиота кишечника и фиброз органов
- Мультиштаммовый пробиотик в терапии неалкогольной жировой болезни печени (НАЖБП)
- Гепатоцеллюлярная карцинома и микробиота кишечника
- Стеатоз печени, ось кишечник-печень и микробиом
- НАЖБП, кишечный микробиом и пробиотики
- НАЖБП, ЛПС и окислительный стресс: нутрицевтические и пробиотические подходы к лечению
- Неалкогольная жировая болезнь печени может контролироваться индолом
- Печень обезжиривают пробиотики
- Лечение неалкогольной жировой болезни печени пробиотиками
- Аутоиммунный гепатит и кишечная микробиота
- Снижение риска гепатокарциномы с помощью лакто- и пропионовокислых бактерий
- Неалкогольная жировая болезнь печени. Взаимосвязь с кишечной микробиотой
- Фруктоза - кишечная проницаемость - НАЖБП
- Кишечные бактерии Klebsiella pneumonia могут вызывать НАЖБП
- Связь между кишечными бактериями и противоопухолевыми иммунными реакциями в печени
Литература
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharm. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Cusi, K. A global view of the interplay between non-alcoholic fatty liver disease and diabetes. Lancet Diabetes Endocrinol. 2022, 10, 284–296. [Google Scholar] [CrossRef]
- Katsarou, A.; Moustakas, I.I.; Pyrina, I.; Lembessis, P.; Koutsilieris, M.; Chatzigeorgiou, A. Metabolic inflammation as an instigator of fibrosis during non-alcoholic fatty liver disease. World J. Gastroenterol. 2020, 26, 1993–2011. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Ni, L.; Zhuge, F.; Fu, Z. The Gut Microbiota and Its Metabolites, Novel Targets for Treating and Preventing Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2020, 64, 2000375. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.-H.; Jin, Z.; Yang, X.-X.; Lou, J.; Shan, W.-X.; Hu, Y.-X.; Du, Q.; Liao, Q.-S.; Xie, R.; Xu, J.-Y. Role of gut microbiota via the gut-liver-brain axis in digestive diseases. World J. Gastroenterol. 2020, 26, 6141–6162. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef]
- Knudsen, C.; Neyrinck, A.M.; Lanthier, N.; Delzenne, N.M. Microbiota and nonalcoholic fatty liver disease: Promising prospects for clinical interventions? Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 393–400. [Google Scholar] [CrossRef]
- Ji, Y.; Yin, Y.; Sun, L.; Zhang, W. The Molecular and Mechanistic Insights Based on Gut-Liver Axis: Nutritional Target for Non-Alcoholic Fatty Liver Disease (NAFLD) Improvement. Int. J. Mol. Sci. 2020, 21, 3066. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.A.; Adegboyega, P.; van Rooijen, N.; Tagalicud, A.; Allman, M.; Wallace, M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J. Hepatol. 2007, 47, 571–579. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.C.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M., Jr.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef]
- Santos-Laso, A.; Gutiérrez-Larrañaga, M.; Alonso-Peña, M.; Medina, J.M.; Iruzubieta, P.; Arias-Loste, M.T.; López-Hoyos, M.; Crespo, J. Pathophysiological Mechanisms in Non-Alcoholic Fatty Liver Disease: From Drivers to Targets. Biomedicines 2021, 10, 46. [Google Scholar] [CrossRef]
- Fuhri Snethlage, C.M.; Nieuwdorp, M.; van Raalte, D.H.; Rampanelli, E.; Verchere, B.C.; Hanssen, N.M.J. Auto-immunity and the gut microbiome in type 1 diabetes: Lessons from rodent and human studies. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101544. [Google Scholar] [CrossRef]
- Hanssen, N.M.J.; de Vos, W.M.; Nieuwdorp, M. Fecal microbiota transplantation in human metabolic diseases: From a murky past to a bright future? Cell Metab. 2021, 33, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Modoux, M.; Rolhion, N.; Mani, S.; Sokol, H. Tryptophan Metabolism as a Pharmacological Target. Trends Pharmacol. Sci. 2021, 42, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Shi, Y.; Chen, C.; Wu, F.; Chen, Z. A narrative review of the roles of indoleamine 2,3-dioxygenase and tryptophan-2,3-dioxygenase in liver diseases. Ann. Transl. Med. 2021, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L.; Henson, J.D.; Hall, S. Intestinal Dysbiosis, the Tryptophan Pathway and Nonalcoholic Steatohepatitis. Int. J. Tryptophan Res. 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Yin, Y.; Li, Z.; Zhang, W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2019, 11, 1712. [Google Scholar] [CrossRef]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. 2009, 2, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. The Uniqueness of Tryptophan in Biology: Properties, Metabolism, Interactions and Localization in Proteins. Int. J. Mol. Sci. 2020, 21, 8776. [Google Scholar] [CrossRef]
- Hyland, N.P.; Cavanaugh, C.R.; Hornby, P.J. Emerging effects of tryptophan pathway metabolites and intestinal microbiota on metabolism and intestinal function. Amino Acids 2022, 54, 57–70. [Google Scholar] [CrossRef]
- Palego, L.; Betti, L.; Rossi, A.; Giannaccini, G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J. Amino Acids 2016, 2016, 8952520. [Google Scholar] [CrossRef]
- Bröer, S.; Bröer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 2017, 474, 1935–1963. [Google Scholar] [CrossRef]
- Ramadan, T.; Camargo, S.M.R.; Herzog, B.; Bordin, M.; Pos, K.M.; Verrey, F. Recycling of aromatic amino acids via TAT1 allows efflux of neutral amino acids via LAT2-4F2hc exchanger. Pflügers Arch.-Eur. J. Physiol. 2007, 454, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S. Adaptation of plasma membrane amino acid transport mechanisms to physiological demands. Pflügers Arch. 2002, 444, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Camargo, S.M.R.; Vuille-dit-Bille, R.N.; Meier, C.F.; Verrey, F. ACE2 and gut amino acid transport. Clin. Sci. 2020, 134, 2823–2833. [Google Scholar] [CrossRef] [PubMed]
- Jando, J.; Camargo, S.M.R.; Herzog, B.; Verrey, F. Expression and regulation of the neutral amino acid transporter B0AT1 in rat small intestine. PLoS ONE 2017, 12, e0184845. [Google Scholar] [CrossRef]
- Jones, S.P.; Guillemin, G.J.; Brew, B.J. The kynurenine pathway in stem cell biology. Int. J. Tryptophan Res. 2013, 6, 57–66. [Google Scholar] [CrossRef]
- Badawy, A.A.B.; Guillemin, G. The Plasma [Kynurenine]/[Tryptophan] Ratio and Indoleamine 2,3-Dioxygenase: Time for Appraisal. Int. J. Tryptophan Res. 2019, 12. [Google Scholar] [CrossRef]
- Verrey, F.; Singer, D.; Ramadan, T.; Vuille-dit-Bille, R.N.; Mariotta, L.; Camargo, S.M.R. Kidney amino acid transport. Pflügers Arch. -Eur. J. Physiol. 2009, 458, 53–60. [Google Scholar] [CrossRef]
- Bröer, S. Amino Acid Transport Across Mammalian Intestinal and Renal Epithelia. Physiol. Rev. 2008, 88, 249–286. [Google Scholar] [CrossRef]
- Badawy, A.A.B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10. [Google Scholar] [CrossRef]
- Rafice, S.A.; Chauhan, N.; Efimov, I.; Basran, J.; Raven, E.L. Oxidation of L-tryptophan in biology: A comparison between tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase. Biochem. Soc. Trans. 2009, 37, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, N.; Watanabe, Y.; Kawanishi, K.; Hashimoto, Y.; Hayaishi, O. Inhibition of indoleamine 2,3-dioxygenase and tryptophan 2,3-dioxygenase by β-carboline and indole derivatives. Arch. Biochem. Biophys. 1984, 232, 602–609. [Google Scholar] [CrossRef]
- King, N.J.C.; Thomas, S.R. Molecules in focus: Indoleamine 2,3-dioxygenase. Int. J. Biochem. Cell Biol. 2007, 39, 2167–2172. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, O. Biochemical and medical aspects of the indoleamine 2,3-dioxygenase-initiated l-tryptophan metabolism. Biochem. Biophys. Res. Commun. 2005, 338, 12–19. [Google Scholar] [CrossRef]
- Pantouris, G.; Serys, M.; Yuasa, H.J.; Ball, H.J.; Mowat, C.G. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino Acids 2014, 46, 2155–2163. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Hunt, N.H.; Ball, H.J. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: Characteristics and potential roles in health and disease. Amino Acids 2013, 45, 1319–1329. [Google Scholar] [CrossRef]
- Marszalek-Grabska, M.; Walczak, K.; Gawel, K.; Wicha-Komsta, K.; Wnorowska, S.; Wnorowski, A.; Turski, W.A. Kynurenine emerges from the shadows–Current knowledge on its fate and function. Pharmacol. Ther. 2021, 225, 107845. [Google Scholar] [CrossRef]
- Phillips, R.S.; Iradukunda, E.C.; Hughes, T.; Bowen, J.P. Modulation of Enzyme Activity in the Kynurenine Pathway by Kynurenine Monooxygenase Inhibition. Front. Mol. Biosci. 2019, 6, 3. [Google Scholar] [CrossRef]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed]
- Banoglu, E.; King, R.S. Sulfation of indoxyl by human and rat aryl (phenol) sulfotransferases to form indoxyl sulfate. Eur. J. Drug Metab. Pharm. 2002, 27, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Schnabl, B. Indoles: Metabolites produced by intestinal bacteria capable of controlling liver disease manifestation. J. Intern. Med. 2019, 286, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.M. The quantitative determination of indolic microbial tryptophan metabolites in human and rodent samples: A systematic review. J. Chromatogr. B 2021, 1186, 123008. [Google Scholar] [CrossRef] [PubMed]
- Koduru, L.; Lakshmanan, M.; Hoon, S.; Lee, D.-Y.; Lee, Y.K.; Ow, D.S.-W. Systems Biology of Gut Microbiota-Human Receptor Interactions: Toward Anti-inflammatory Probiotics. Front. Microbiol 2022, 13, 846555. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Dong, F.; Perdew, G.H. The aryl hydrocarbon receptor as a mediator of host-microbiota interplay. Gut Microbes 2020, 12, 1859812. [Google Scholar] [CrossRef]
- Disner, G.R.; Lopes-Ferreira, M.; Lima, C. Where the Aryl Hydrocarbon Receptor Meets the microRNAs: Literature Review of the Last 10 Years. Front. Mol. Biosci. 2021, 8, 725044. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Stephens, G.L.; Wang, Q.; Swerdlow, B.; Bhat, G.; Kolbeck, R.; Fung, M. Kynurenine 3-monooxygenase mediates inhibition of Th17 differentiation via catabolism of endogenous aryl hydrocarbon receptor ligands. Eur. J. Immunol. 2013, 43, 1727–1734. [Google Scholar] [CrossRef]
- Illés, P.; Krasulová, K.; Vyhlídalová, B.; Poulíková, K.; Marcalíková, A.; Pečinková, P.; Sirotová, N.; Vrzal, R.; Mani, S.; Dvořák, Z. Indole microbial intestinal metabolites expand the repertoire of ligands and agonists of the human pregnane X receptor. Toxicol. Lett. 2020, 334, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Vilarem, M.J.; Maurel, P.; Pascussi, J.M. Xenoreceptors CAR and PXR Activation and Consequences on Lipid Metabolism, Glucose Homeostasis, and Inflammatory Response. Mol. Pharm. 2008, 5, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tabb, M.M.; Nelson, E.L.; Grün, F.; Verma, S.; Sadatrafiei, A.; Lin, M.; Mallick, S.; Forman, B.M.; Thummel, K.E.; et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Investig. 2006, 116, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Sun, S.; Wang, P.; Sun, Y.; Hu, Q.; Wang, X. The Mechanism of Secretion and Metabolism of Gut-Derived 5-Hydroxytryptamine. Int. J. Mol. Sci. 2021, 22, 7931. [Google Scholar] [CrossRef]
- Koopman, N.; Katsavelis, D.; Hove, A.S.T.; Brul, S.; Jonge, W.J.d.; Seppen, J. The Multifaceted Role of Serotonin in Intestinal Homeostasis. Int. J. Mol. Sci. 2021, 22, 9487. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Grifka-Walk, H.M.; Jenkins, B.R.; Kominsky, D.J. Amino Acid Trp: The Far Out Impacts of Host and Commensal Tryptophan Metabolism. Front. Immunol. 2021, 12, 2005. [Google Scholar] [CrossRef] [PubMed]
- Guzel, T.; Mirowska-Guzel, D. The Role of Serotonin Neurotransmission in Gastrointestinal Tract and Pharmacotherapy. Molecules 2022, 27, 1680. [Google Scholar] [CrossRef]
- Zahar, S.; Schneider, N.; Makwana, A.; Chapman, S.; Corthesy, J.; Amico, M.; Hudry, J. Dietary tryptophan-rich protein hydrolysate can acutely impact physiological and psychological measures of mood and stress in healthy adults. Nutr. Neurosci. 2022, 1–10. [Google Scholar] [CrossRef]
- Acuña-Castroviejo, D.; Escames, G.; Venegas, C.; Díaz-Casado, M.E.; Lima-Cabello, E.; López, L.C.; Rosales-Corral, S.; Tan, D.-X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef]
- Sui, G.; Jia, L.; Quan, D.; Zhao, N.; Yang, G. Activation of the gut microbiota-kynurenine-liver axis contributes to the development of nonalcoholic hepatic steatosis in nondiabetic adults. Aging 2021, 13, 21309–21324. [Google Scholar] [CrossRef] [PubMed]
- Nagano, J.; Shimizu, M.; Hara, T.; Shirakami, Y.; Kochi, T.; Nakamura, N.; Ohtaki, H.; Ito, H.; Tanaka, T.; Tsurumi, H.; et al. Effects of Indoleamine 2,3-Dioxygenase Deficiency on High-Fat Diet-Induced Hepatic Inflammation. PLoS ONE 2013, 8, e73404. [Google Scholar] [CrossRef]
- Vivoli, E.; Cappon, A.; Cozzi, A.; Navari, N.; Gargano, M.; Fallarino, F.; Marra, F. A novel role for the kynurenine pathway in experimental steatohepatitis. Dig. Liver Dis. 2015, 47, e21. [Google Scholar] [CrossRef]
- Rojas, I.Y.; Moyer, B.J.; Ringelberg, C.S.; Wilkins, O.M.; Pooler, D.B.; Ness, D.B.; Coker, S.; Tosteson, T.D.; Lewis, L.D.; Chamberlin, M.D.; et al. Kynurenine-Induced Aryl Hydrocarbon Receptor Signaling in Mice Causes Body Mass Gain, Liver Steatosis, and Hyperglycemia. Obesity 2021, 29, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, B.; Hu, Y.; Zhao, Y. New Insights Into Gut-Bacteria-Derived Indole and Its Derivatives in Intestinal and Liver Diseases. Front. Pharmacol. 2021, 12, 769501. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Yanagi, K.; Cheng, C.; Alaniz, R.C.; Lee, K.; Jayaraman, A. Interactions between gut microbiota and non-alcoholic liver disease: The role of microbiota-derived metabolites. Pharmacol. Res. 2019, 141, 521–529. [Google Scholar] [CrossRef]
- Zhou, D.; Fan, J.-G. Microbial metabolites in non-alcoholic fatty liver disease. World J. Gastroenterol. 2019, 25, 2019–2028. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Lai, J.K.L.; Qiao, L.; Fan, J.G. Role of gut microbial metabolites in nonalcoholic fatty liver disease. J. Dig. Dis. 2019, 20, 181–188. [Google Scholar] [CrossRef]
- Beaumont, M.; Neyrinck, A.M.; Olivares, M.; Rodriguez, J.; de Rocca Serra, A.; Roumain, M.; Bindels, L.B.; Cani, P.D.; Evenepoel, P.; Muccioli, G.G.; et al. The gut microbiota metabolite indole alleviates liver inflammation in mice. FASEB J. 2018, 32, 6681–6693. [Google Scholar] [CrossRef]
- Knudsen, C.; Neyrinck, A.M.; Leyrolle, Q.; Baldin, P.; Leclercq, S.; Rodriguez, J.; Beaumont, M.; Cani, P.D.; Bindels, L.B.; Lanthier, N.; et al. Hepatoprotective Effects of Indole, a Gut Microbial Metabolite, in Leptin-Deficient Obese Mice. J. Nutr. 2021, 151, 1507–1516. [Google Scholar] [CrossRef]
- Ma, L.; Li, H.; Hu, J.; Zheng, J.; Zhou, J.; Botchlett, R.; Matthews, D.; Zeng, T.; Chen, L.; Xiao, X.; et al. Indole Alleviates Diet-Induced Hepatic Steatosis and Inflammation in a Manner Involving Myeloid Cell 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3. Hepatology 2020, 72, 1191–1203. [Google Scholar] [CrossRef]
- Ji, Y.; Gao, Y.; Chen, H.; Yin, Y.; Zhang, W. Indole-3-Acetic Acid Alleviates Nonalcoholic Fatty Liver Disease in Mice via Attenuation of Hepatic Lipogenesis, and Oxidative and Inflammatory Stress. Nutrients 2019, 11, 2062. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Namkung, J.; Hwang, I.; Kim, H.; Lim, A.; Park, H.J.; Lee, H.W.; Han, K.-H.; Park, S.; Jeong, J.-S.; et al. Serotonin signals through a gut-liver axis to regulate hepatic steatosis. Nat. Commun. 2018, 9, 4824. [Google Scholar] [CrossRef]
- Zhao, Z.-H.; Xin, F.-Z.; Xue, Y.; Hu, Z.; Han, Y.; Ma, F.; Zhou, D.; Liu, X.-L.; Cui, A.; Liu, Z.; et al. Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Sehgal, R.; Ilha, M.; Vaittinen, M.; Kaminska, D.; Männistö, V.; Kärjä, V.; Tuomainen, M.; Hanhineva, K.; Romeo, S.; Pajukanta, P.; et al. Indole-3-Propionic Acid, a Gut-Derived Tryptophan Metabolite, Associates with Hepatic Fibrosis. Nutrients 2021, 13, 3509. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Sun, C.; Chen, Y.; Du, F.; Yang, Y.; Wu, G. Indole-3-propionic Acid-aggravated CCl(4)-induced Liver Fibrosis via the TGF-β1/Smads Signaling Pathway. J. Clin. Transl. Hepatol. 2021, 9, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Wegermann, K.; Howe, C.; Henao, R.; Wang, Y.; Guy, C.D.; Abdelmalek, M.F.; Diehl, A.M.; Moylan, C.A. Serum Bile Acid, Vitamin E, and Serotonin Metabolites Are Associated With Future Liver-Related Events in Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2021, 5, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Kyritsi, K.; Chen, L.; O’Brien, A.; Francis, H.; Hein, T.W.; Venter, J.; Wu, N.; Ceci, L.; Zhou, T.; Zawieja, D.; et al. Modulation of the Tryptophan Hydroxylase 1/Monoamine Oxidase-A/5-Hydroxytryptamine/5-Hydroxytryptamine Receptor 2A/2B/2C Axis Regulates Biliary Proliferation and Liver Fibrosis During Cholestasis. Hepatology 2020, 71, 990–1008. [Google Scholar] [CrossRef]
- Ko, M.; Kamimura, K.; Owaki, T.; Nagoya, T.; Sakai, N.; Nagayama, I.; Niwa, Y.; Shibata, O.; Oda, C.; Morita, S.; et al. Modulation of serotonin in the gut-liver neural axis ameliorates the fatty and fibrotic changes in non-alcoholic fatty liver. Dis. Model. Mech. 2021, 14, dmm048922. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, G.; Bai, J.; Zhao, N.; Wang, Q.; Zhou, R.; Li, G.; Hu, C.; Li, X.; Tao, K.; et al. Role of Indole-3-Acetic Acid in NAFLD Amelioration After Sleeve Gastrectomy. Obes. Surg. 2021, 31, 3040–3052. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Yang, L.; Li, Y.; Chen, J.; Zhang, X.; Wang, H.; Zhai, S.; Jiang, X.; Meca, G.; Wang, S.; et al. Melatonin alleviates Ochratoxin A-induced liver inflammation involved intestinal microbiota homeostasis and microbiota-independent manner. J. Hazard. Mater. 2021, 413, 125239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yan, A.; Liu, X.; Ma, Y.; Zhao, F.; Wang, M.; Loor, J.J.; Wang, H. Melatonin ameliorates ochratoxin A induced liver inflammation, oxidative stress and mitophagy in mice involving in intestinal microbiota and restoring the intestinal barrier function. J. Hazard. Mater. 2021, 407, 124489. [Google Scholar] [CrossRef] [PubMed]
- Sewerynek, E.; Melchiorri, D.; Reiter, R.J.; Ortiz, G.G.; Lewinski, A. Lipopolysaccharide-induced hepatotoxicity is inhibited by the antioxidant melatonin. Eur. J. Pharmacol. Environ. Toxicol. Pharmacol. 1995, 293, 327–334. [Google Scholar] [CrossRef]
- Bellanti, F.; Villani, R.; Facciorusso, A.; Vendemiale, G.; Serviddio, G. Lipid oxidation products in the pathogenesis of non-alcoholic steatohepatitis. Free Radic. Biol. Med. 2017, 111, 173–185. [Google Scholar] [CrossRef]
- Celinski, K.; Konturek, P.C.; Slomka, M.; Cichoz-Lach, H.; Brzozowski, T.; Konturek, S.J.; Korolczuk, A. Effects of treatment with melatonin and tryptophan on liver enzymes, parameters of fat metabolism and plasma levels of cytokines in patients with non-alcoholic fatty liver disease—14 months follow up. J. Physiol. Pharm. 2014, 65, 75–82. [Google Scholar]
- Ayyash, A.; Holloway, A.C. Fluoxetine-induced hepatic lipid accumulation is mediated by prostaglandin endoperoxide synthase 1 and is linked to elevated 15-deoxy-Δ12,14PGJ2. J. Appl. Toxicol. 2021, 42, 1004–1015. [Google Scholar] [CrossRef]
- Ayyash, A.; Holloway, A.C. Fluoxetine-induced hepatic lipid accumulation is linked to elevated serotonin production. Can. J. Physiol. Pharmacol. 2021, 99, 983–988. [Google Scholar] [CrossRef]
- Li, R.; Zhu, W.; Huang, P.; Yang, Y.; Luo, F.; Dai, W.; Shen, L.; Pei, W.; Huang, X. Olanzapine leads to nonalcoholic fatty liver disease through the apolipoprotein A5 pathway. Biomed. Pharmacother. 2021, 141, 111803. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and inflammation—Story of a double-edged blade. J. Pineal Res. 2018, 65, e12525. [Google Scholar] [CrossRef]
- Kanova, M.; Kohout, P. Tryptophan: A Unique Role in the Critically Ill. Int. J. Mol. Sci. 2021, 22, 11714. [Google Scholar] [CrossRef]
- Fiore, A.; Murray, P.J. Tryptophan and indole metabolism in immune regulation. Curr. Opin. Immunol. 2021, 70, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Grondin, J.A.; Khan, W.I. Tryptophan-derived serotonin-kynurenine balance in immune activation and intestinal inflammation. FASEB J. 2021, 35, e21888. [Google Scholar] [CrossRef] [PubMed]
- Borghi, M.; Pariano, M.; Solito, V.; Puccetti, M.; Bellet, M.M.; Stincardini, C.; Renga, G.; Vacca, C.; Sellitto, F.; Mosci, P.; et al. Targeting the Aryl Hydrocarbon Receptor With Indole-3-Aldehyde Protects From Vulvovaginal Candidiasis via the IL-22-IL-18 Cross-Talk. Front. Immunol. 2019, 10, 2364. [Google Scholar] [CrossRef]
- Tuomainen, M.; Lindström, J.; Lehtonen, M.; Auriola, S.; Pihlajamäki, J.; Peltonen, M.; Tuomilehto, J.; Uusitupa, M.; de Mello, V.D.; Hanhineva, K. Associations of serum indolepropionic acid, a gut microbiota metabolite, with type 2 diabetes and low-grade inflammation in high-risk individuals. Nutr. Diabetes 2018, 8, 4983. [Google Scholar] [CrossRef] [PubMed]
- Abedi, S.; Vessal, M.; Asadian, F.; Takhshid, M.A. Association of serum kynurenine/tryptophan ratio with poor glycemic control in patients with type2 diabetes. J. Diabetes Metab. Disord. 2021, 20, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Scarale, M.G.; Mastroianno, M.; Prehn, C.; Copetti, M.; Salvemini, L.; Adamski, J.; De Cosmo, S.; Trischitta, V.; Menzaghi, C. Circulating Metabolites Associate with and Improve the Prediction of All-Cause Mortality in Type 2 Diabetes. Diabetes 2022, 71, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Raynal, S.; Bailbé, D.; Gausseres, B.; Carbonne, C.; Autier, V.; Movassat, J.; Kergoat, M.; Portha, B. Expression of the kynurenine pathway enzymes in the pancreatic islet cells. Activation by cytokines and glucolipotoxicity. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 980–991. [Google Scholar] [CrossRef]
- Lin, H.V.; Efanov, A.M.; Fang, X.; Beavers, L.S.; Wang, X.; Wang, J.; Gonzalez Valcarcel, I.C.; Ma, T. GPR142 Controls Tryptophan-Induced Insulin and Incretin Hormone Secretion to Improve Glucose Metabolism. PLoS ONE 2016, 11, e0157298. [Google Scholar] [CrossRef]
- Cussotto, S.; Delgado, I.; Anesi, A.; Dexpert, S.; Aubert, A.; Beau, C.; Forestier, D.; Ledaguenel, P.; Magne, E.; Mattivi, F.; et al. Tryptophan Metabolic Pathways Are Altered in Obesity and Are Associated With Systemic Inflammation. Front. Immunol. 2020, 11, 557. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Hazlett, H.F.; Nemani, K.V.; Trask, H.W.; West, R.J.; Lupien, L.E.; Collins, A.J.; Ringelberg, C.S.; et al. Inhibition of the aryl hydrocarbon receptor prevents Western diet-induced obesity. Model for AHR activation by kynurenine via oxidized-LDL, TLR2/4, TGFβ, and IDO1. Toxicol. Appl. Pharmacol. 2016, 300, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Laurans, L.; Venteclef, N.; Haddad, Y.; Chajadine, M.; Alzaid, F.; Metghalchi, S.; Sovran, B.; Denis, R.G.P.; Dairou, J.; Cardellini, M.; et al. Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health. Nat. Med. 2018, 24, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Gáspár, R.; Halmi, D.; Demján, V.; Berkecz, R.; Pipicz, M.; Csont, T. Kynurenine Pathway Metabolites as Potential Clinical Biomarkers in Coronary Artery Disease. Mult. Implic. Kynurenine Pathw. Inflamm. Dis. Diagn. Ther. Appl. 2022, 12. [Google Scholar] [CrossRef]
- Melhem, N.J.; Taleb, S. Tryptophan: From Diet to Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 9904. [Google Scholar] [CrossRef]
- Song, P.; Ramprasath, T.; Wang, H.; Zou, M.-H. Abnormal kynurenine pathway of tryptophan catabolism in cardiovascular diseases. Cell. Mol. Life Sci. 2017, 74, 2899–2916. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ye, D.; Wang, Z.; Pan, H.; Lu, X.; Wang, M.; Xu, Y.; Yu, J.; Zhang, J.; Zhao, M.; et al. The Role of Interleukin-6 Family Members in Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 9, 818890. [Google Scholar] [CrossRef]
- Cason, C.A.; Dolan, K.T.; Sharma, G.; Tao, M.; Kulkarni, R.; Helenowski, I.B.; Doane, B.M.; Avram, M.J.; McDermott, M.M.; Chang, E.B.; et al. Plasma microbiome-modulated indole- and phenyl-derived metabolites associate with advanced atherosclerosis and postoperative outcomes. J. Vasc. Surg. 2018, 68, 1552–1562.e1557. [Google Scholar] [CrossRef]
- Kappel, B.A.; De Angelis, L.; Heiser, M.; Ballanti, M.; Stoehr, R.; Goettsch, C.; Mavilio, M.; Artati, A.; Paoluzi, O.A.; Adamski, J.; et al. Cross-omics analysis revealed gut microbiome-related metabolic pathways underlying atherosclerosis development after antibiotics treatment. Mol. Metab. 2020, 36, 100976. [Google Scholar] [CrossRef]
- Boulet, L.; Flore, P.; Le Gouellec, A.; Toussaint, B.; Pépin, J.L.; Faure, P. Is tryptophan metabolism involved in sleep apnea-related cardiovascular co-morbidities and cancer progression? Med. Hypotheses 2015, 85, 415–423. [Google Scholar] [CrossRef]
- İriz, A.; Şemsi, R.; Eser, B.; Arslan, B.; Dinçel, A.S. The evaluation of serum tryptophan and kynurenine levels in patients with obstructive sleep apnea syndrome. Sleep Breath. 2021, 25, 1389–1398. [Google Scholar] [CrossRef]
